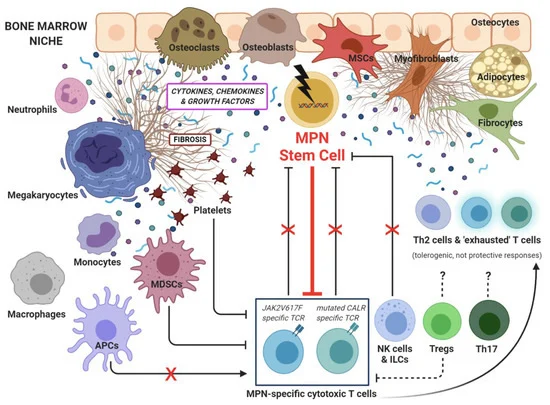
Hematological malignancies have perplexed physicians and patients alike for decades. Recent strides in understanding the intricate molecular circuitry governing hematopoiesis have begun shedding light on a particularly puzzling group of blood cancers known as myeloproliferative neoplasms (MPNs). These stem cell-derived clonal disorders wreak havoc in the bone marrow, dysregulating blood cell production and releasing immature myeloid cells into circulation . The iconic Philadelphia chromosome negative MPNs include polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), and chronic myelogenous leukemia (CML) . Today I’ll be focusing on unraveling the intricacies of PV, PMF, and CML - an assorted yet interconnected trifecta of myeloproliferative madness!
In PV, a single mutated hematopoietic stem cell begins overproducing red blood cells, causing the blood to thicken and dramatically elevating the risk of clots. Identifying the JAK2V617F mutation unlocked our understanding of overactive JAK-STAT signaling as the culprit behind this erythrocytosis. Yet 30% of PV patients lack this mutation, kindling ongoing investigations seeking alternative genetic explanations.
Whereas PV primarily disrupts red blood cell equilibrium, PMF stems from malignant stem cells that fibroticize bone marrow and skew megakaryocyte and granulocyte production . The pro-fibrotic cytokine TGF-β likely orchestrates this starter cell transformation and subsequent scar tissue accumulation . Recurrent mutations in JAK2, CALR, and MPL implicate aberrant JAK-STAT, MAPK, and PI3K signaling downstream .
Unlike its Philadelphia chromosome negative counterparts, CML’s hallmark is the BCR-ABL1 fusion gene originating from a chromosomal translocation between chromosomes 9 and 22. This unique swap generates a hyperactive tyrosine kinase driving unrestrained myeloproliferation. Yet with tyrosine kinase inhibitors (TKIs) like imatinib demonstrating durable cytogenetic responses, CML has transitioned into a chronic, manageable illness for many patients .
Despite their differences, intersecting signals unite this MPN triad. JAK-STAT pathways potently stimulate myeloproliferation across PV, PMF, and CML . PCA analysis even clustered these diseases together, affirming a shared transcriptional signature. Ongoing efforts probing inflammatory signaling and clonal architecture continue unraveling new connections. These insights illuminate promising therapeutic inroads for tailored MPN treatment based on a mutation’s downstream effects rather than its simple presence.
Yet MPNs remain complex beasts, with our grasp of their intricacies far from absolute. Their heterogeneous nature hints at an interplay between constitutional genetics and environmental exposures in disease pathogenesis. Could microbial, dietary, or other lifestyle factors influence MPN susceptibility and progression? How do aging stem cells and clonal hematopoiesis factor into the equation? Much remains enshrouded in mystery. Unraveling the true triggers and biology of MPNs will be no simple feat. But the threads we continue disentangling inch us ever closer to definitive solutions for MPN patients worldwide.
References:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5472936/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6378940/
https://www.nejm.org/doi/full/10.1056/nejmoa051113
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6298360/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5503459/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6311211/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6378940/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6765346/
https://www.nejm.org/doi/full/10.1056/NEJMoa022240
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6378940/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6378940/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5472936/

